
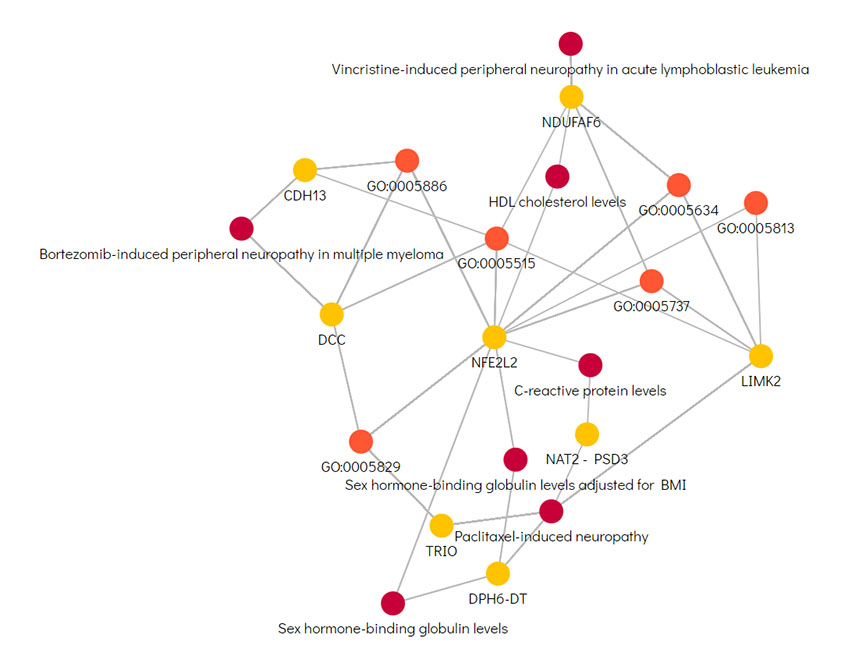
JuvAI Biology
LLM-enabled biomedical knowledge graph for optimal target and indication selection
JuvAI Chemistry
State-of-the-art AI/ML platform for molecular design and optimization
JuvAI Clinic
Cutting-edge ML tools for adaptive clinical trials and precision patient selection


08.03.2021
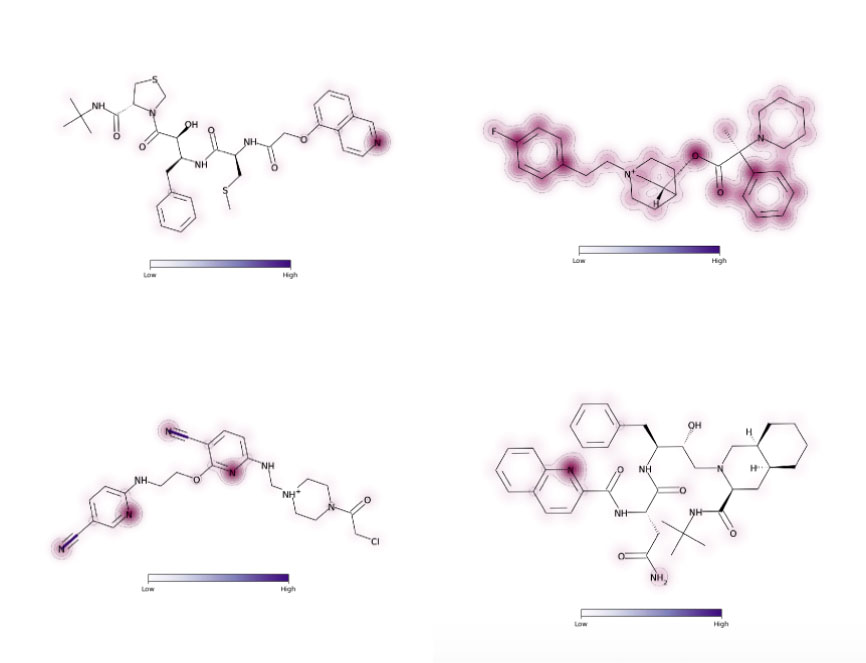
11.16.2021
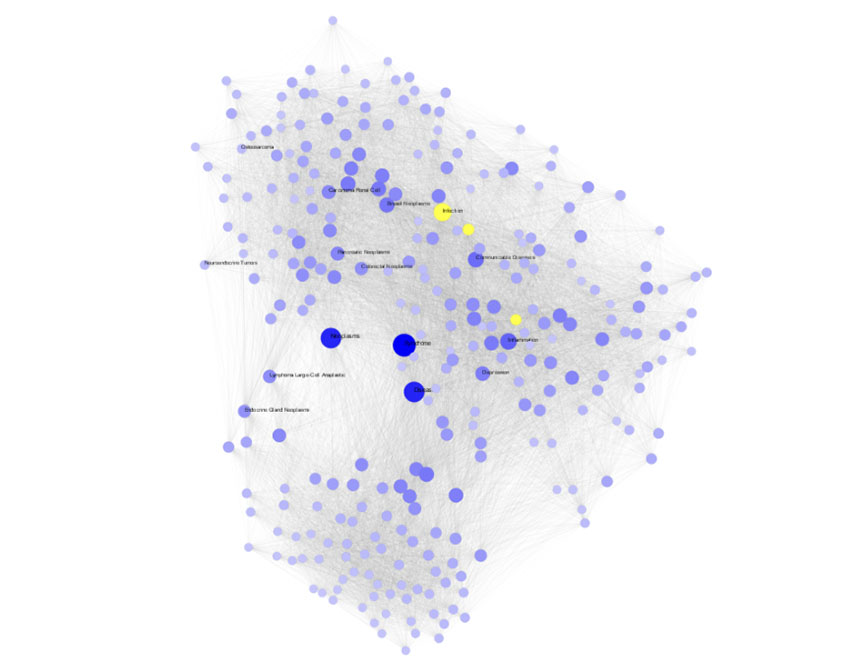
22.02.2022
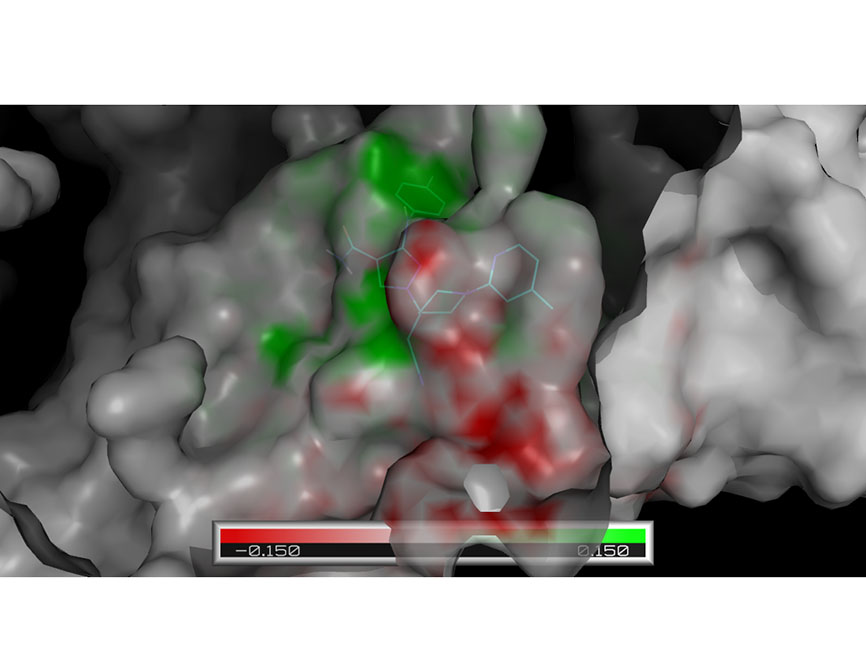
22.02.2022
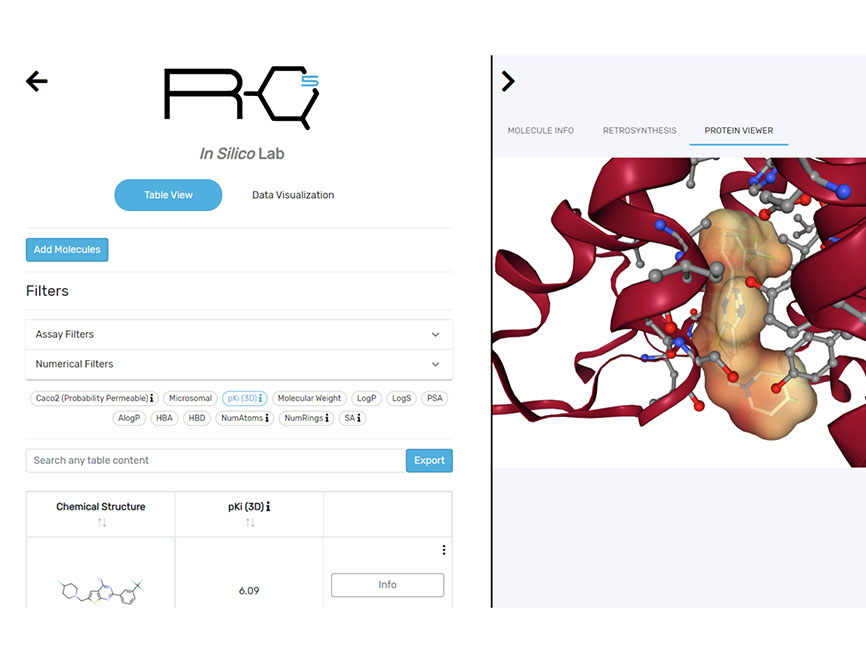
22.02.2022
01.03.2022
01.03.2022
07.06.2022